
在实验室美美摸鱼突然被微信消息打断,原来是导师发来一篇微信公众号里的Selenium教程,要求我用爬虫为药材的质谱数据获取参考化学成分表,于是有了这一篇笔记。
PubChem
正好刚刚学过 Selenium,当时我没有多想就开干了,复盘时才意识到这是一个非常错误的决定。导师的研究药物主要是石斛,我就到 PubChem 上搜索 Dendrobium Nobile, 果然在 Taxonomy 里有这一条目。往下翻一翻,发现 Chemicals and Bioactivities 的列表可以下载。点击下载,美美收工,继续摸鱼。
下载后发现没这么简单。 pubchem_taxid_xxxxx_consolidatedcompoundtaxonomy.csv 的文件里只有化合物名,编号和源数据库链接。对于质谱数据分析,我至少需要化合物的化学式和分子质量。仔细一看链接,发现石斛的数据来源主要是以下三个数据库:
这正是爬虫的应用场景。我只需要对每种化合物,依次访问对应的数据库条目,并记录他的化学式和分子质量。由于 PubChem 将 Metabolites 和 Natural Products 下载为了两个单独的 .csv 表格,先写一个简单的脚本将他们合并为一个:
#!/usr/bin/env python3import osimport sysimport pandas as pd
def main(): input_files = ['metabolites.csv', 'natural_products.csv'] dfs = []
for fname in input_files:
# Load and validate each input file if not os.path.isfile(fname): sys.exit(f"Input file not found: {fname}")
# Read only the necessary columns from the CSV df = pd.read_csv( fname, usecols=['Compound_CID', 'Compound', 'Source_Chemical', 'Source_Chemical_URL'] ) dfs.append(df)
# Concatenate both DataFrames into a single one combined = pd.concat(dfs, ignore_index=True) # Remove duplicate entries based on Compound_CID, keeping the first occurrence combined = combined.drop_duplicates(subset=['Compound_CID'], keep='first')
output_file = 'pubchem_combined.csv' combined.to_csv(output_file, index=False) print(f"Merged CSV written to: {output_file}")
if __name__ == "__main__": main()Selenium
爬虫脚本的思路还是比较简单的:
- 将上一步准备好的
.csv文件作为输入,其中每行都包含一个指向对应数据库条目的Source_Chemical_URL列。 - 对于每个 URL,借助 XPath 使用特定于站点的 parser 从页面的 HTML 结构中提取所需的数据。
- 模块化,通过一个 dispatcher 根据域名(NPASS、Knapsack 或 Wikidata)调用不同的 parser。
- 解析后,结果被写回
.csv表格,附加两列:分子质量和化学式。
基本配置
首先是配置 ChromeDriver:
def setup_driver(headless=False):
# Finds the chromedriver executable in your system chromedriver_path = shutil.which("chromedriver") if not chromedriver_path: sys.exit("ERROR: chromedriver executable not found in PATH.")
# Sets up Chrome options (headless if requested) options = webdriver.ChromeOptions() options.page_load_strategy = 'eager' if headless: options.add_argument('--headless=new') options.add_argument('--disable-gpu')
# Returns a webdriver.Chrome instance service = Service(chromedriver_path) driver = webdriver.Chrome(service=service, options=options) # driver.set_page_load_timeout(PAGE_LOAD_TIMEOUT) # npass websites are really slow return driver之后设置参数。这里我将超时设置为 30 秒,每次爬取的间隔设置为 1 秒。由于 NPASS 的网站加载较慢,其实需要更长的超时时间。在实际运行中我直接去掉了 TimeoutException。
PAGE_LOAD_TIMEOUT = 30 # how long to wait for a page to loadDEFAULT_PAUSE = 1.0 # seconds to wait between scrapes to avoid overloading serversParsers
Knapsack
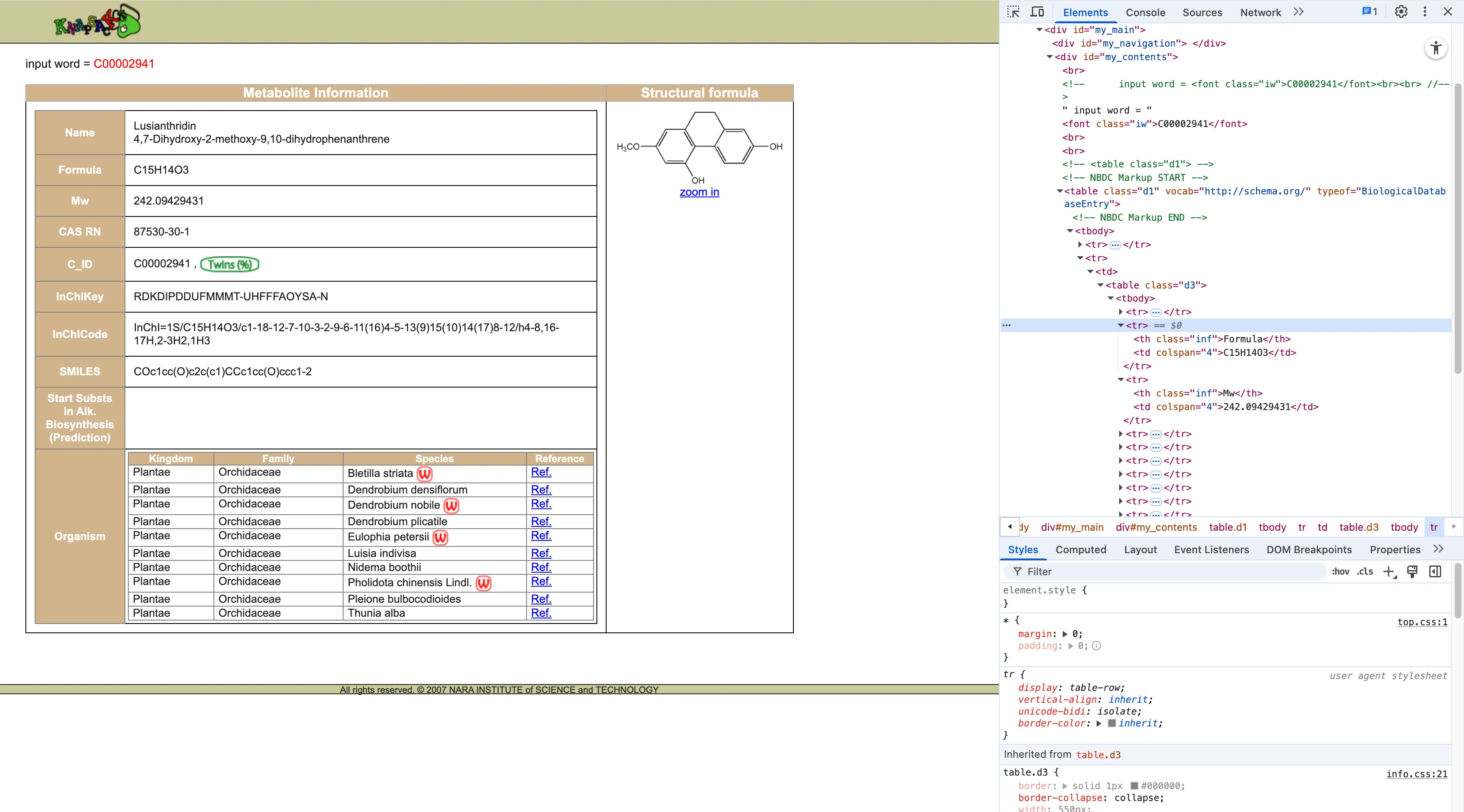
Knapsack 以这样的结构存储数据:
<tr> <th class="inf">Formula</th> <td colspan="4">C15H14O3</td></tr><tr> <th class="inf">Mw</th> <td colspan="4">242.09429431</td></tr>数据以「结构化的行」形式存储。<tr> 元素中, <th> 作为标签, <td> 作为值。这样我可以写一个简单的 get_text_label_in_table 的 helper 可靠地匹配 <th> 中的文本(如「Formula」、「Mw」),然后获取相邻的 <td>。
代码如下:
# Looks in a table for a row with the given label and gets the corresponding value from the same row# Find any table row <tr> where the first cell (whether it's a <th> or <td>) exactly matches the label.def get_text_label_in_table(driver, label): try: row = driver.find_element( By.XPATH, f"//table//tr[normalize-space(.//th[1] | .//td[1])='{label}']" ) return row.find_element(By.XPATH, './td[1]').text.strip() except NoSuchElementException: return None
# Parser for Knapsack# Extracts Formula and Mw from chemical entry pages on knapsackfamily.com using table-based scrapingdef parse_knapsack(driver): formula = get_text_label_in_table(driver, 'Formula') weight = get_text_label_in_table(driver, 'Mw') if weight is None: weight = get_text_label_in_table(driver, 'Molecular weight') return weight, formulaNPASS
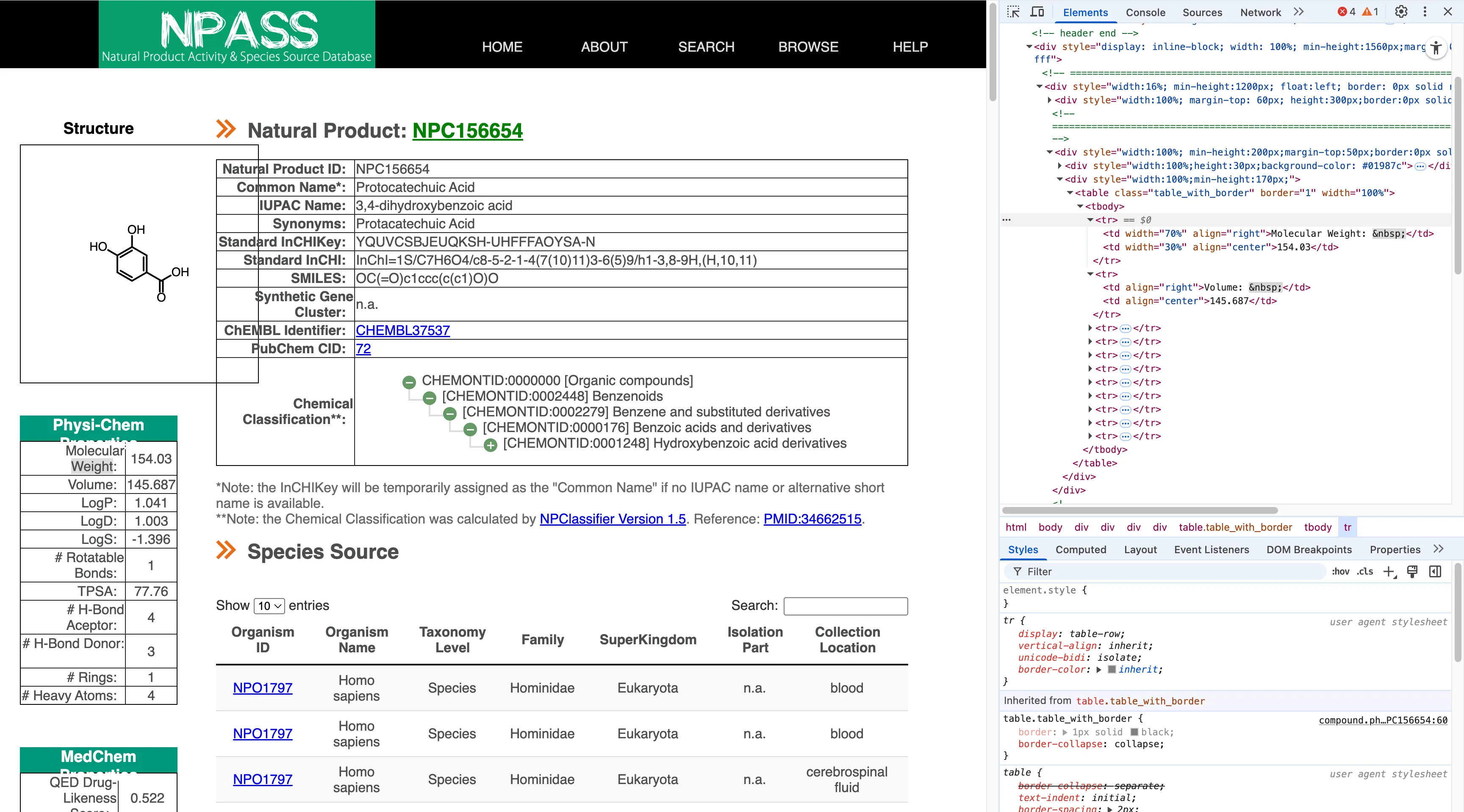
<tr> <td width="70%" align="right">Molecular Weight: </td> <td width="30%" align="center">154.03</td></tr>NPASS 相比之下就复杂一些。首先,NPASS有时会使用像 <dt>/<dd>(定义列表)这样的定义标签。但在其他情况下,他们又会使用没有 <th> 标签,直接将文字保存在 <td> 标签的普通表格。因此我需要跟多的判断逻辑:
- 首先尝试
<dt>/<dd>查找(首选方法)。 - 如果未找到,我就直接寻找
td[1][contains(normalize-space(.),'Molecular Weight')]之后的下一个<td>元素里保存的值。
代码如下:
# Parser for NPASS# Extracts Formula and Molecular Weight from npass.bidd.group using <dt>/<dd> tags and fallbacks to table parsing if neededdef parse_npass(driver):
# Try extracting formula from <dt>/<dd> try: formula = driver.find_element( By.XPATH, "//dt[contains(normalize-space(),'Molecular Formula')]/following-sibling::dd[1]" ).text.strip() except NoSuchElementException: formula = None
# Try extracting weight from <dt>/<dd> try: weight = driver.find_element( By.XPATH, "//dt[contains(normalize-space(),'Molecular Weight')]/following-sibling::dd[1]" ).text.strip() except NoSuchElementException: weight = None
# NPASS often uses a <table class="table_with_border">…</table> for Mw; # if the dt/dd lookup failed or returned '0', fall back to grabbing from the table. if not weight or weight == '0': try: weight = driver.find_element( By.XPATH, "//table[contains(@class,'table_with_border')]" "//tr[td[1][contains(normalize-space(.),'Molecular Weight')]]/td[2]" ).text.strip() except NoSuchElementException: weight = None
return weight, formulaWikidata
当我为 Wikidata 写 parser 的时候,发现 Wikidata 有提供 API,并有 wbgetentities 函数。这样就可以直接通过 https://www.wikidata.org/w/api.php?action=wbgetentities&ids=QXXX&props=claims&format=json ,直接获得一个干净的 JSON 文件:
{ "entities": { "Qxxx": { "claims": { "P274": [...], // Formula "P2067": [...] // Molecular weight } } }}这样就只需要从 JSON 文件中读取值就好了,代码如下:
# Parser for Wikidata# Uses the Wikidata API to extract:# P274: chemical formula# P2067: molecular weight# Handles potential nested dictionary responsesdef parse_wikidata(entity_id): import urllib.request, json
api_url = ( 'https://www.wikidata.org/w/api.php' '?action=wbgetentities&ids=%s&props=claims&format=json' % entity_id ) try: with urllib.request.urlopen(api_url, timeout=PAGE_LOAD_TIMEOUT) as f: data = json.load(f) claims = data['entities'][entity_id]['claims'] formula = None weight = None if 'P274' in claims: formula = claims['P274'][0]['mainsnak']['datavalue']['value'] if 'P2067' in claims: weight = claims['P2067'][0]['mainsnak']['datavalue']['value'] # Wikidata returns a dict {'amount': '+<value>', 'unit': ...}; extract the numeric amount if isinstance(weight, dict): raw_amount = weight.get('amount') weight = raw_amount.lstrip('+') if raw_amount is not None else None return weight, formula except Exception as e: print(f"WARNING: failed to fetch Wikidata {entity_id}: {e}", file=sys.stderr) return None, None在这里我就意识到用爬虫直接爬去 ui 界面上的内容可能是一个非常糟糕的选择,本文后面的部分会继续提到。
Dispatcher
在 parser 都完成后,我还需要一个 dispatcher,根据 URL 所指向的网站域名来选择正确的 parser,结构非常简单:
# Dispatcher: Determine Which Parser to Usedef dispatch_parse(driver, url): hostname = urlparse(url).hostname or '' if 'knapsackfamily.com' in hostname: driver.get(url) return parse_knapsack(driver) if 'bidd.group' in hostname: driver.get(url) return parse_npass(driver) if 'wikidata.org' in hostname: entity_id = url.rstrip('/').rsplit('/', 1)[-1] return parse_wikidata(entity_id) print(f"WARNING: no parser available for {url}", file=sys.stderr) return None, NoneCLI
我希望我的脚本能够作为一个通用的工具,于是简单写了一个命令行界面,主要有 4 个参数:input_csv, output_csv, --headless, --pause。
# Command-line Interfaceif __name__ == '__main__': p = argparse.ArgumentParser( description='Scrape molecular weight and formula for pubchem chemicals.' ) p.add_argument('input_csv', help='Input CSV (final_pubchem.csv)') p.add_argument('output_csv', help='Output CSV with Mw and Formula') p.add_argument('--headless', action='store_true', help='Run Chrome in headless mode') p.add_argument('--pause', type=float, default=DEFAULT_PAUSE, help='Seconds to pause between requests') args = p.parse_args() main(args.input_csv, args.output_csv, args.pause, args.headless)PUG REST API
前面说过,我在写 Wikidata 的爬虫时就意识到,对于「获取参考化学成分表」这一任务来说,爬虫这个方法有点过于不优雅了。很快我就了解到原来 PubChem 提供了 API 平台:PUG REST,而且并不鼓励直接使用爬虫大量爬取网页内容。我立刻决定弥补我犯下的错误,重写整个脚本。
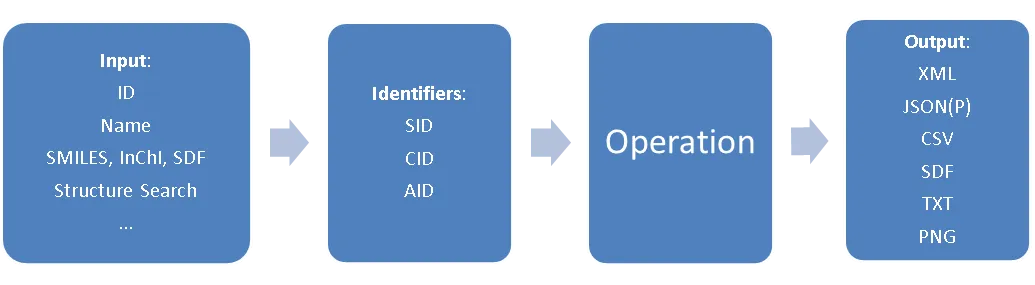
PUG REST 的查询都基于 PubChem 编号,即 SID 代表物质编号,CID 代表成分编号,AID 代表检验编号。想要查询某个编号的物质 / 成分 / 检验相关的信息,就可以使用这样的 URL 结构:
https://pubchem.ncbi.nlm.nih.gov/rest/pug /compound/name/vioxx /property/InChI /TXT prolog input operation output
同时还支持大量的输出格式:
Output Format Description XML standard XML, for which a schema is available JSON JSON, JavaScript Object Notation JSONP JSONP, like JSON but wrapped in a callback function ASNB standard binary ASN.1, NCBI’s native format in many cases ASNT NCBI’s human-readable text flavor of ASN.1 SDF chemical structure data CSV comma-separated values, spreadsheet compatible PNG standard PNG image data TXT plain text
这样一来,对于我的需求,我就可以通过这个 API 平台快速的批量查询 CID 列表,同时还可以自定义查询的内容。这需要调取这样的一条 URL:
url = f"https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/{namespace}/property/{props}/JSON"其中 namespace 是一个以 「 , 」分隔的 CID 列表,而 props 则可以填入任意一个或多个 PUG REST 支持的查询条目。
这样就可以写出一个用于查询的 helper:
payload = {namespace: ",".join(ids), "property": props}backoff = 1.0
for attempt in range(retries): try: r = SESSION.post(url, data=payload, timeout=30) r.raise_for_status() data = r.json()["PropertyTable"]["Properties"] key_field = "CID" if namespace == "cid" else "Name" return {str(item[key_field]): item for item in data} except Exception as exc: if attempt == retries - 1: raise time.sleep(backoff) backoff *= 2 continue对一个包含一列 CID 信息的化合物表格(如开头用脚本合并得到的表格),我就可以快速批量的获取他们的化学式和分子质量,并将新信息分别以单独的列加入回表格中:
df = pd.read_csv(in_path)if args.id_column not in df.columns: sys.exit(f"Column {args.id_column!r} not found in {in_path}")
# ensure CID keys are clean strings without '.0'if args.cid: ids = df[args.id_column].astype(int).astype(str).tolist()else: ids = df[args.id_column].astype(str).tolist()namespace = "cid" if args.cid else "name"
# load cache if presentcache: Dict[str, Dict[str, str]] = {}if cache_path: cache = load_cache(cache_path)
# figure out which IDs still need queryingto_query = [i for i in ids if i not in cache]print(f"{len(ids)} total IDs / {len(to_query)} to query (cached {len(ids)-len(to_query)})")
# batch loopfor i in range(0, len(to_query), args.batch_size): batch = to_query[i : i + args.batch_size] print(f"Fetching batch {i // args.batch_size + 1} (size {len(batch)}) ...", end="", flush=True) try: props_dict = pug_request(namespace, batch, args.props) cache.update(props_dict) print(" done.") except Exception as exc: print(f" failed ({exc}).") time.sleep(args.sleep)
# save cacheif cache_path: save_cache(cache, cache_path) # optionally remove cache file after run if args.auto_delete_cache and cache_path.exists(): cache_path.unlink() print("Deleted cache", cache_path)
# add columns back to DataFrameprop_names = args.props.split(",")for prop in prop_names: key_series = ( df[args.id_column].astype(int).astype(str) if args.cid else df[args.id_column].astype(str) ) df[prop] = key_series.map(lambda x: cache.get(x, {}).get(prop, ""))使用 API 的运行速度比爬虫快的多,爬虫加载一个网页的时间已经够 API 查询上百条化合物,工作效率得到了巨大的增加。同时还不需要担心大量检索触发数据库网站的访问限制,以及部分数据库的 ui 界面加载速度较慢造成超时报错。
在这之后我还想进一步简化流程,希望能够将从 PubChem 手动下载化合物列表这一步也自动化,实现输入 Taxonomy ID,直接输出对应的,包含所有所需信息的化合物表格,但是这遇到了一些困难。PUG REST 并没有将 Taxonomy ID 和 CID 直接联系在一起的功能,而最接近的只有 Taxonomy ID -> AID -> CID。文档中有写到:
Assays and Bioactivities
The following operation returns a list of compounds involved in a given taxonomy. Valid output formats are XML, JSON(P), ASNT/B, and TXT.
https://pubchem.ncbi.nlm.nih.gov/rest/pug/taxonomy/taxid/2697049/aids/TXT
There is no operation available to directly retrieve the bioactivity data associated with a given taxonomy, as often the data volume is huge. However, one can first get the list of AIDs using the above link, and then aggregate the concise bioactivity data from each AID, e.g.:
https://pubchem.ncbi.nlm.nih.gov/rest/pug/assay/aid/1409578/concise/JSON
实际操作后发现,对于大量的 taxonomy,尤其是我的项目所涉及的天然中药材,/taxonomy/taxid/xxxxxxx/aids/ 都只会返回 404,即数据库内没有对应的 Taxonomy ID -> AID 的记录,这样也就没有办法继续从 AID 列表获取 CID 了。
质谱软件
在花费大量时间靠着自己那一点点业余计算机知识为各种数据库构建 parser 并尝试自动化收集数据后,我才开始考虑是否有人之前做过类似的事情,但做得远比我更好。于是我简单搜索就发现了这份质谱软件列表。质谱分析问题已经有着一个完整的工具生态系统,并且很多工具非常适配我的需求,甚至有更高级的工具利用机器学习算法直接通过质谱波峰预测蛋白质序列。
比如,在华盛顿大学上学的朋友和我提到他们学校开发的 Crux 工具,里面的很多方法完全可以更快更好的完成我的任务:
tide-index Create an index of all peptides in a fasta file, for use in subsequent calls to tide-search.
tide-search Search a collection of spectra against a sequence database, provided either as a FASTA file or an index, returning a collection of peptide-spectrum matches (PSMs). This is a fast search engine, but it runs most quickly if provided with a peptide index built with tide-index.
comet Search a collection of spectra against a sequence database, returning a collection of PSMs. This search engine runs directly on a protein database in FASTA format.
percolator Re-rank and assign confidence estimates to a collection of PSMs using the Percolator algorithm. Optionally, also produce protein rankings using the Fido algorithm.
kojak Search a collection of spectra against a sequence database, finding cross-linked peptide matches.
还有很多利用相似算法的商业程序,有着完整的前端甚至 web 界面,比如 InstaNovo 等,只要上传光谱文件,就能直接得到 Transformer 模型的预测结果。
总结
捣鼓了半天原来在重复造轮子确实很让我失望,但我并不后悔自己编写脚本,因为这我理解了幕后机制,也是我第一次爬虫实战。我犯的最大错误是没有先思考为什么,就直接去考虑怎么做,过于专注于「爬虫」这一技术而不是「分析质谱数据」这一目的。在科学计算领域,往往站在巨人的肩膀上会更有效。